


Signal detection
The entry into force of this Regulation on 28 January 2022 brought a change of approach to pharmacovigilance which is now based on a signal detection process. This system allows active and continuous monitoring of pharmacovigilance data in order to detect potential safety problems early.
In pharmacovigilance, a “signal” refers to information from one or more sources that suggests a possible new causal association or a new aspect of a known causal association between a drug and an adverse event.
This process includes steps for the detection, prioritisation, validation and evaluation of identified signals in order to take appropriate management measures (amendment of SPC, communication on a possible risk, suspension of marketing authorisation...).
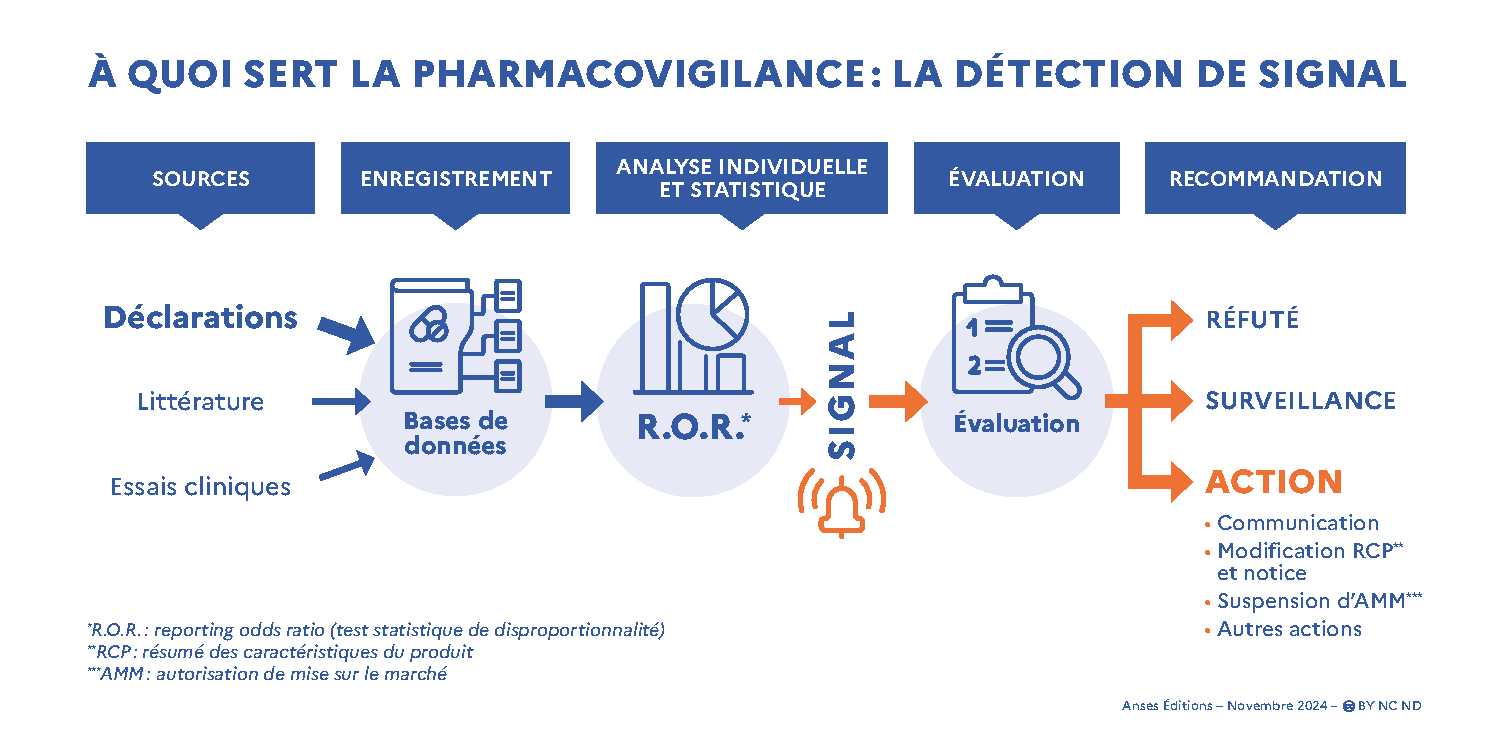
How are signals detected and treated in pharmacovigilance?
The data sources that give rise to signals are multiple. Although data from clinical trials and literature are also taken into account, spontaneous statements are the majority source of these signals.
Signal detection from spontaneous reports is done both through individual analysis of the received cases and through an analysis of aggregated data in pharmacovigilance databases. Statistical analysis (based on the calculation of the Reporting Odds Ratio or ROR) makes it possible to detect possible statistical associations between clinical signs and drugs in the databases. The clinical relevance of these associations requires further validation by an analysis of the cases concerned.
The signals thus detected are only hypotheses, which then need to be confirmed and then evaluated to determine whether risk management measures are necessary. This analysis aims to determine whether a causal link with the drug is possible, in which case actions will be to be studied, or whether it can be ruled out. Where the available data do not make it possible to conclude, additional information, in particular by means of new declarations, is necessary. The signal is then “under surveillance”.
A signal detection carried out at European and national level
Regulation 2019/6 requires MAHs to implement a continuous process of detection and monitoring of signals for their medicinal products at European level. Statistical analyses are thus regularly carried out by manufacturers for their medicinal products from the Eudravigilance veterinary database, which brings together all reported cases of pharmacovigilance for all medicinal products authorised in Europe.
All the signals detected are recorded on a platform managed by the European Medicines Agency (List of signals from Veterinary Signal Management · IRIS (europa.eu)). For each signal the medicinal product concerned, the clinical signs observed and the affected species are mentioned. It also includes the actions proposed by the MAHs following this detection (rebuttal signal, under supervision, modification of the SmPC and package leaflet of the medicinal product, other risk management measures...). The MA changes proposed by the holders are then assessed by the competent authorities. At the same time, the authorities also monitor pharmacovigilance data for certain products/product families/substances on the basis of a risk analysis. Finally, the conclusions on the benefit-risk balance of each medicinal product are also recorded on this IRIS platform, which are recorded by the MA holder at least once a year.
In France, a statistical analysis (via a calculation of the MMR) is also carried out every two months by the ANMV on the basis of all the cases recorded in the national database of veterinary pharmacovigilance, in order to identify the potential signals from adverse events occurring in the national territory. The signals thus detected can then be reported at European level, and possibly lead to risk management measures such as amendments to marketing authorisations or communication actions by the ANMV.
Learn more about actions related to signal detection in veterinary pharmacovigilance
For more information on changes to SmPC related to pharmacovigilance, for centralised marketing authorisations, the list of regulatory recommendations related to EMA pharmacovigilance.