


Détection de signal
L’entrée en vigueur de ce règlement le 28 janvier 2022 a apporté un changement d’approche en matière de pharmacovigilance qui est maintenant basée sur un processus de détection de signal. Ce système permet une surveillance active et continue des données de pharmacovigilance afin de détecter précocement des problèmes de sécurité potentiels.
En pharmacovigilance, un « signal » désigne des informations provenant d'une ou de plusieurs sources qui suggèrent une possible nouvelle association causale ou un nouvel aspect d'une association causale connue entre un médicament et un événement indésirable.
Ce processus comprend des étapes de détection, de hiérarchisation, de validation et d’évaluation des signaux identifiés afin de prendre des mesures de gestion appropriées (modification de RCP, communication sur un risque éventuel, suspension d’AMM…).
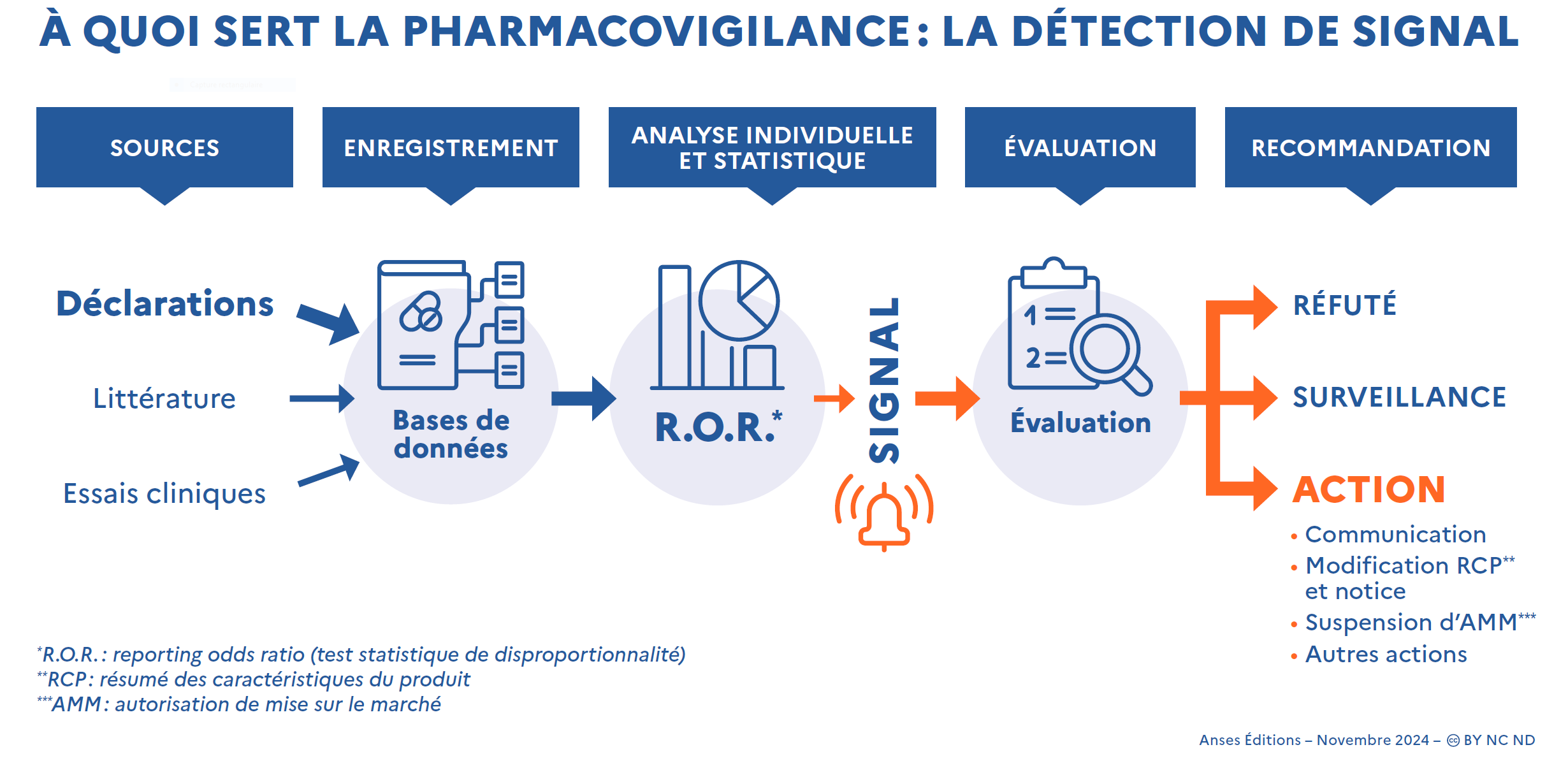
Comment sont détectés et traités les signaux en pharmacovigilance ?
Les sources de données à l’origine de signaux sont multiples. Même si les données issues des essais cliniques et de la littérature sont également prises en compte, les déclarations spontanées constituent la source majoritaire de ces signaux.
La détection de signal à partir des déclarations spontanées se fait à la fois via l’analyse individuelle des cas reçus et via une analyse des données agrégées dans les bases de données de pharmacovigilance. Une analyse statistique (basée sur le calcul du Reporting Odds Ratio ou ROR) permet de détecter d’éventuelles associations statistiques entre des signes cliniques et des médicaments dans les bases de données. La pertinence clinique de ces associations nécessite d’être ensuite validée par une analyse des cas concernés.
Les signaux ainsi détectés ne sont que des hypothèses, qui nécessitent ensuite d’être confirmées puis évaluées pour déterminer si des mesures de gestion de risque sont nécessaires. Cette analyse vise à établir si un lien de causalité avec le médicament est possible, auquel cas des actions seront à étudier, ou s’il peut être écarté. Lorsque les données disponibles ne permettent pas de conclure, l’obtention d’informations complémentaires, notamment par le biais de nouvelles déclarations, est nécessaire. Le signal est alors « sous surveillance ».
Une détection de signal effectuée au niveau Européen et national
Le règlement 2019/6 impose aux titulaires d’AMM de mettre en œuvre un processus continu de détection et de suivi des signaux pour leurs médicaments au niveau européen. Des analyses statistiques sont ainsi régulièrement effectuées par les industriels pour leurs médicaments à partir de la base de données Eudravigilance veterinary qui réunit tous les cas de pharmacovigilance déclarés pour l’ensemble des médicaments autorisés en Europe.
L’ensemble des signaux ainsi détectés est enregistré sur une plateforme gérée par l’Agence Européenne du Médicament (List of signals from Veterinary Signal Management · IRIS (europa.eu)). Pour chaque signal sont mentionnés le médicament concerné, les signes cliniques observés et l’espèce affectée. On y retrouve également les actions proposées par les titulaires d’AMM suite à cette détection (signal réfuté, sous surveillance, modification du RCP et de la notice du médicament, autres mesures de gestion du risque…). Les modifications d’AMM proposées par les titulaires font ensuite l’objet d’une évaluation par les autorités compétentes. En parallèle, les autorités réalisent de leur côté une surveillance des données de pharmacovigilance pour certains produits/familles de produits/substances sur la base d’une analyse de risque. Enfin, sont également enregistrées sur cette plateforme IRIS, les conclusions sur le rapport bénéfice/risque de chaque médicament, qui sont consignées par le titulaire de l’AMM au moins une fois par an.
En France, une analyse statistique (via un calcul du ROR) est également effectuée tous les deux mois par l’ANMV à partir de l’ensemble des cas enregistrés dans la base de données nationale de pharmacovigilance vétérinaire, afin d’identifier les signaux potentiels issus des évènements indésirables survenus sur le territoire national. Les signaux ainsi détectés peuvent ensuite faire l’objet de signalements au niveau européen, et éventuellement aboutir à des mesures de gestion de risque telles que des modifications d’AMM ou des actions de communication de la part de l’ANMV.
En savoir plus sur les actions liées à la détection de signal en pharmacovigilance vétérinaire
Pour plus d’informations concernant les modifications de RCP en lien avec la pharmacovigilance, consulter la Lettre d’information mensuelle sur les médicaments vétérinaires | Anses - Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail ou, pour les AMM centralisées, la liste des recommandations réglementaires liées à la pharmacovigilance de l’EMA.
Retrouvez également nos alertes et communiqués en pharmacovigilance vétérinaire : Pharmacovigilance | Anses - Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail
Pour recevoir l’actualité relative aux médicaments vétérinaires, abonnez-vous à la newsletter sur L’actualité du médicament vétérinaire (Abonnés ANMV (anses.fr)